Marvelous Misadventures in Bioinformatics
A blog on some snippets of my work in bioinformatics. Hopefully you find something useful here and avoid stupid mistakes I made.
Visualisation of protein domains architecture
Representing protein domain architecture is often used in research, especially when investigating unknown sequences.
They usually look like this.
However, the DOG1.0 appears to deprecated. IBS2.0 is an now available plotter for biological sequences, not just protein domains.
Apart from manually drawing it by hand, you could programmatically and dynamically make these plots. However, currently available tools is limited to the R programming language.
drawProteins and seqvisr are two of such tools. I will be covering drawProteins and how to customise the plots by using the BoNT/A (accession: Q7B8V4) from C. botulinum as an example.
Prerequisite
- R (>=4.3.3)
testing on older versions worked without issues, but is not guaranteed
Installation
if (!require("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install("drawProteins")
Usage
-
Load the packages
library(drawProteins) library(ggplot2) #optional -
Load in the data.
By default, drawProteins can load uniprot to load the information. You will able to directly use the information from the annotation
json_dat <- get_features("Q7B8V4") df_dat <- feature_to_dataframe(json_dat)alternatively, load it in one go with
df_dat <- feature_to_dataframe(getfeatures("Q7B8V4")) -
Intialise the ggplot canvas
p <- draw_canvas(df_dat) -
Plot the chains and domains
p <- draw_chains(x, df_dat) p <- draw_domains(x, df_dat) -
Optional: Modify the plot the remove background and change the theme, along with setting the font face. You will need ggplot2 imported
p + theme_bw(base_size = 20, base_family = "Times New Roman" ) theme(panel.grid.minor=element_blank(), panel.grid.major=element_blank()) + theme(axis.ticks = element_blank(), axis.text.y = element_blank()) + theme(panel.border = element_blank()) + theme(legend.title = element_text(size = 20))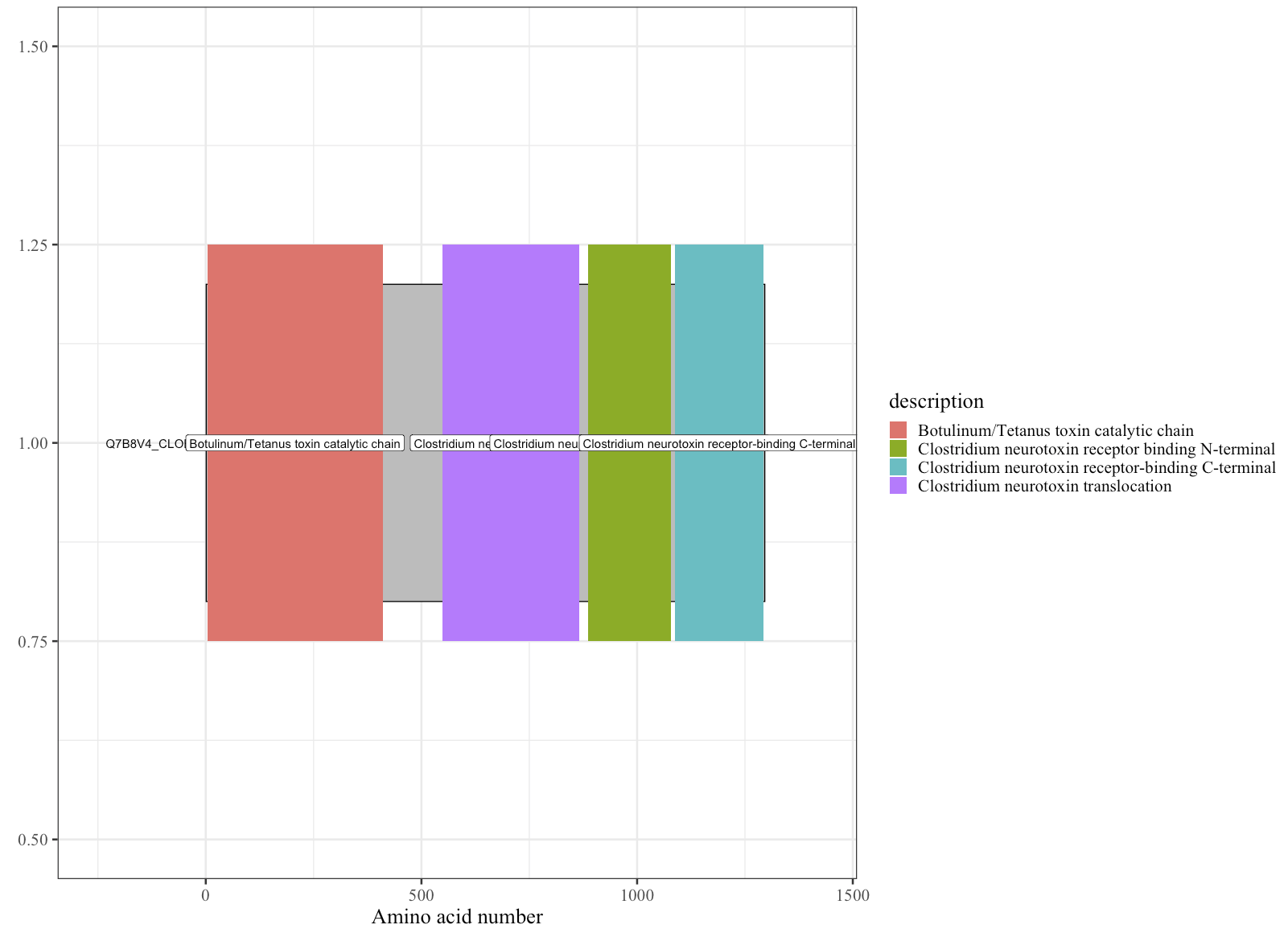
Note: the documentation is lacking, and doesn’t cover other aspects of modfiying the plot
Remove the description on the domains by adding
label_domains = FALSEto draw_domainsp <- draw_domains(x, df_dat, label_domains = FALSE)Change the legend title and key size
p <- p + guides(fill=guide_legend(title="Features", keywidth = unit(2, 'cm')))
Set a colour palaette for your domains, using the okabe colour blind friendly set as example
okabe <- c("#D55E00", "#CC79A7", "#E69F00", "#009E73", "#0072B2") p <- p + scale_fill_manual(values = okabe)
Note that the accession id on the left side is not
Times new roman. This is because thedraw_chainscallsggplotto annotate the file beforehand, it escapes thetheme_bw(base_family=font)override command. We want define a new function, saydraw_chains_timesasdraw_chains_times <- function (p, data = data, outline = "black", fill = "grey", label_chains = TRUE, labels = data[data$type == "CHAIN", ]$entryName, size = 0.5, label_size = 4) { begin = end = NULL p <- p + geom_rect(data = data[data$type == "CHAIN", ], mapping = aes(xmin = begin, xmax = end, ymin = order - 0.2, ymax = order + 0.2), colour = outline, fill = fill, size = size) if (label_chains == TRUE) { p <- p + annotate("text", x = -10, y = data[data$type == "CHAIN", ]$order, label = labels, hjust = 1, size = label_size, family = "Times") } return(p) }Note: if you have already imported
ggplot2, do not addggplot2::in front ofgeom_rectandannotatein the custom functionNow intsead of calling
draw_chains, calldraw_chains_timesor any other name you have defined in this custom function. -
You can plot custom proteins in case Uniprot annotation is not up to your taste/not available.
This is done by injecting a dataframe in this format:
rowname type description begin end length accession entryName taxid order 1 CHAIN dummy 1 1000 1000 test_dummy test_data 1 1 2 DOMAIN dummy domain 1 10 20 10 dummy domain dummy domain 1 1 3 DOMAIN dummy domain 2 40 50 10 dummy domain dummy domain 1 1 this represents a protein of size 1000, with two domains at 10-20 aa and 40-50 aa. I have placed taxid as 1 as placeholder
Assuming your data is in a file called
dummy_data.txtin the same current working directory.library(data.table) df_dat <- data.frame(fread("./dummy_data.txt"), row.names = 1)row.names = 1means the first column is the row names.Now plot as you would in the previous example.
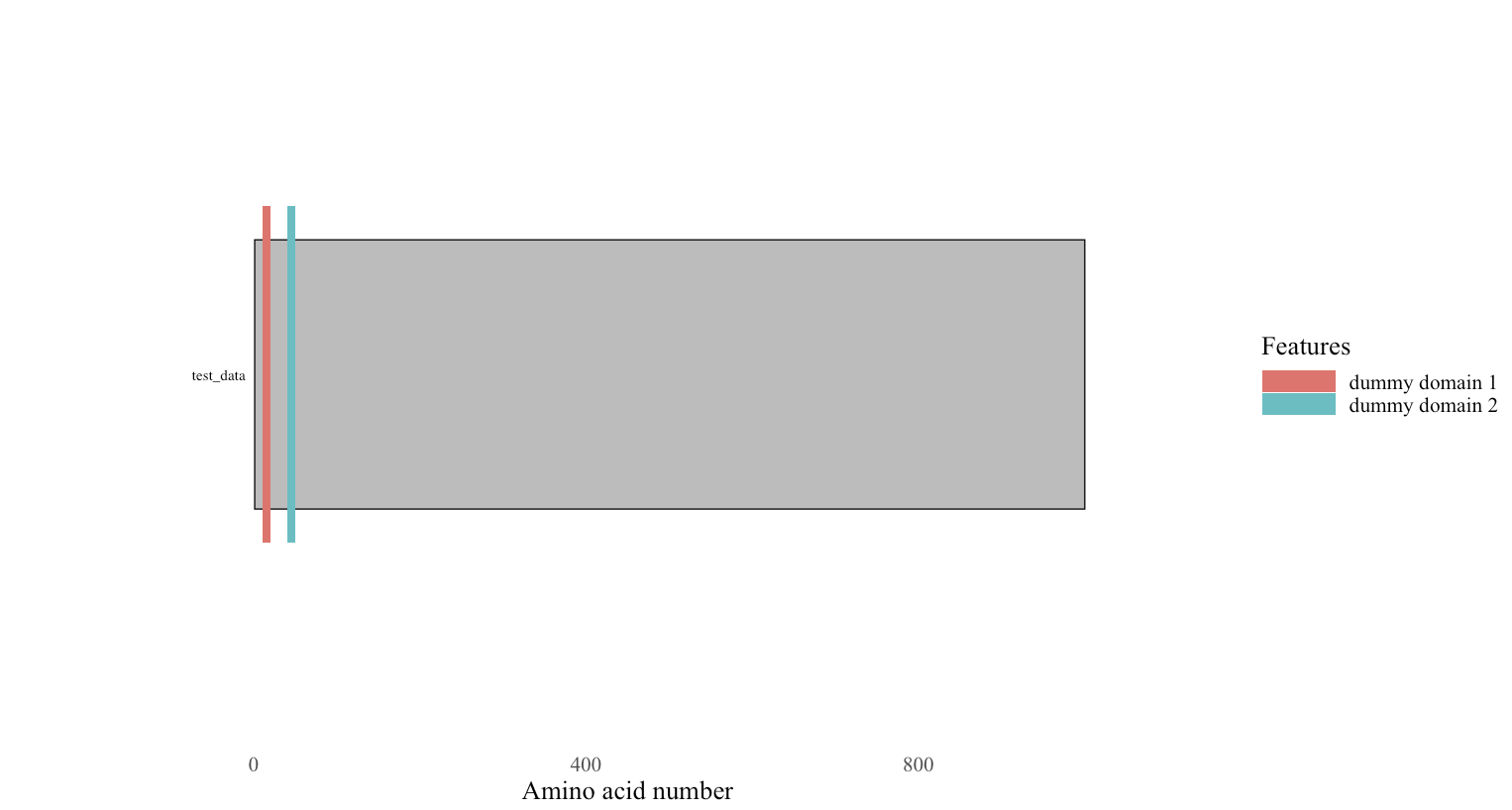
This can be extended to plot multiple proteins.
rowname type description begin end length accession entryName taxid order 1 CHAIN dummy 1 1000 1000 test_dummy test_data 1 1 2 DOMAIN dummy domain 1 10 20 10 dummy domain dummy domain 1 1 3 DOMAIN dummy domain 2 40 50 10 dummy domain dummy domain 1 1 4 CHAIN dummy 1 1000 1000 test_dummy test_data2 1 2 5 DOMAIN dummy domain 1 10 20 10 dummy domain dummy domain 1 2 6 DOMAIN dummy domain 2 40 50 10 dummy domain dummy domain 1 2 By adding to the
orderrow, you can plot multiple proteins.Note: plots are made from bottom up. Keep that in mind.
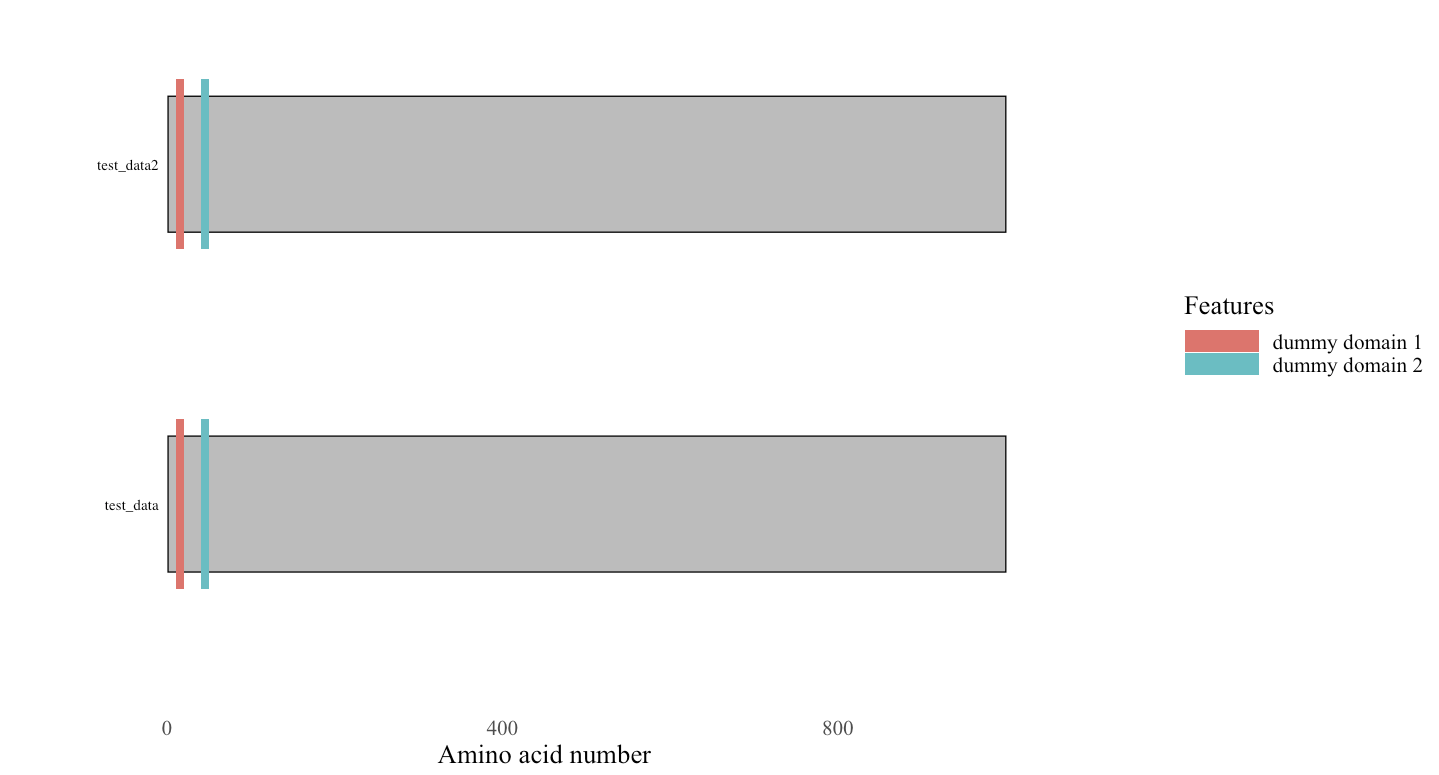
Reference
- https://f1000research.com/articles/7-1105/v1