Marvelous Misadventures in Bioinformatics
A blog on some snippets of my work in bioinformatics. Hopefully you find something useful here and avoid stupid mistakes I made.
Protein Language Models
This is a gentle introduction and tutorial into Protein language models (PLMs). PLMs are large language models pre-trained on vast amounts of protein sequences, usually in a unsupervised fashion where no labels are given to the sequences. The model is tasked to discover and learn the semantics of the protein sequences, effectively understanding the language of proteins (i.e. amino acids).
This tutorial will be covering the ESM2 PLM 650 million parameter model developed by Facebook AI Research. This tutorial shows how to extract the embeddings, a feature rich vector that describes the protein. There are lots of other PLMs available such as ProtBert. I will be embedding the same Gyrase A (Accession: P9WG47) in previous posts as an example.
Prerequisite
- tensorflow
- pytorch
- SeqIO
- transformer
- numpy
- ESM2 installed locally somewhere
Installation
- Get ESM2 here
-
SeqIO
pip install Bio -
Numpy ( numpy should be installed during your tensorflow/pytorch installation )
pip install numpy -
transformers
pip install transformers
Usage
-
import required packages
import os os.environ['TF_CPP_MIN_LOG_LEVEL'] = '3' #this is optional import tensorflow as tf from transformers import AutoTokenizer, TFEsmModel import numpy as np from Bio import SeqIONote: importing
osandos.environ['TF_CPP_MIN_LOG_LEVEL'] = '3'is optional as it only silences the warning logs generated -
Define a function that replaces
B, Z, J, U,andOamino acids from your sequence into the unknown amino acidX.This is because ESM (and to a wider extent, other PLMS) works with the 20 common amino acids, uncommon amino acids may cause problems.
def formatseq(seq): seq = seq.replace("B", "X").replace("Z", "X").replace("J", "X").replace("U", "X").replace("O", "X") return seq -
Set your environmental variables and intialise instance of the ESM2 model and tokenizer
modelDir = "PATH/TO/ESM2/INSTALLATION" tokenizer = AutoTokenizer.from_pretrained(modelDir) model = TFEsmModel.from_pretrained(modelDir) test_sequence = "./P9WG47.fa" #your sequence fasta file -
Initialise an empty list to hold all your sequence strings.
seqs = [] -
Open the fasta file using
Bio.SeqIOand append the strings into the empty list.with open(test_sequence, "r") as readfile: for record in SeqIO.parse(readfile, "fasta"): seqs.append(str(record.seq))Note: SeqIO.parse returns a
Seqobject when you runRecord.seq; adding astr()will force it to be a regular string. -
Remap the uncommon acids (if any) to
Xseqs = [formatseq(seq) for seq in seqs]I know this is not the best or most pythonic way to map stuff, but it works.
-
Tokenize the sequence, feed it into the ESM2 model and retrieve the last hidden state. This is the embedding layer.
inputs = tokenizer(seqs, return_tensors="tf", padding= True, truncation= True, max_length = 1024) #return tf for tensorflow outputs = model(inputs) last_hidden_states = outputs.last_hidden_state per_residue_embeddings = np.array(last_hidden_states)This will yield a np array of size num_of_sequence * length_of_protein * 1280
Note: ESM2 and its other sister models have the same input sequence limit of 1024 amino acids.
Note2: Failure to comply to this 1024 aa limit may poison your GPU. You have been warned
-
Optional: Get the per protein embeddings
Get the per protein emebeddings by averaging over the length of the sequence. This is dimension 1 of the numpy array.
#get per protein embeddings per_protein_embeddings = np.mean(per_residue_embeddings, axis=1) -
View the embeddings and its shape.
Running
#view shapes print(per_residue_embeddings.shape) print(per_protein_embeddings.shape)Will yield a result of
(1, 840, 1280)and(1, 1280)respectively.Running
print(per_protein_embeddings[0]) print(per_residue_embeddings[0])Will yield
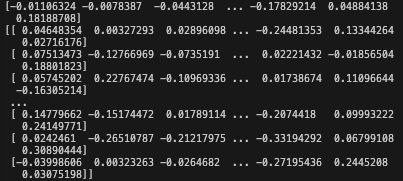
This numerical vector are the embeddings (both per residue, and per protein). They can be used as downstream features for model building or visualisation.
Reference
- https://www.science.org/doi/10.1126/science.ade2574