Marvelous Misadventures in Bioinformatics
A blog on some snippets of my work in bioinformatics. Hopefully you find something useful here and avoid stupid mistakes I made.
Visualisation of phylogenetic tree using ggtree
“But the difficulty is not nearly so great as it at first appears: all this beautiful work can be shown, I think, to follow from a few very simple instincts” - Charles Darwin, 24 November 1859, On the Origin of Species
Following up from inferrence of phylogenetic tree from this tutorial, this tutorial will show how the tree can be visualised using R using ggtree and ggtreeExtra.
Prerequisite
- R
- ggplot2
- ggtree
- ggtreeExtra
- phangorn
- treeio
- ggnewscale
Installation
install.packages("ggplot2")
install.packages("phangorn")
BiocManager::install("ggtree")
BiocManager::install("ggtreeExtra")
BiocManager::install("treeio")
Usage
-
Import packages
library(ggplot2) library(ggtree) library(treeio) library(ggtreeExtra) library(phangorn) library(ggnewscale) -
Import tree file
Assuming your tree file is in the same working directory with the name
virus.newick.virustree <- read.newick("./virus.newick") -
Optional: Get the midpoint of the tree
If you want to midpoint root your tree. Do the following.
y <- midpoint(virustree) getRoot(y) -
Plot the (rerooted) tree
p <- ggtree(y, color = "blue", layout = "rectangular")The layout parameter defaults to
"rectangular", change this to"circular"for larger datasets. -
Add tree tips
p2 <- p + geom_tiplab()Visualise the tree
plot(p2)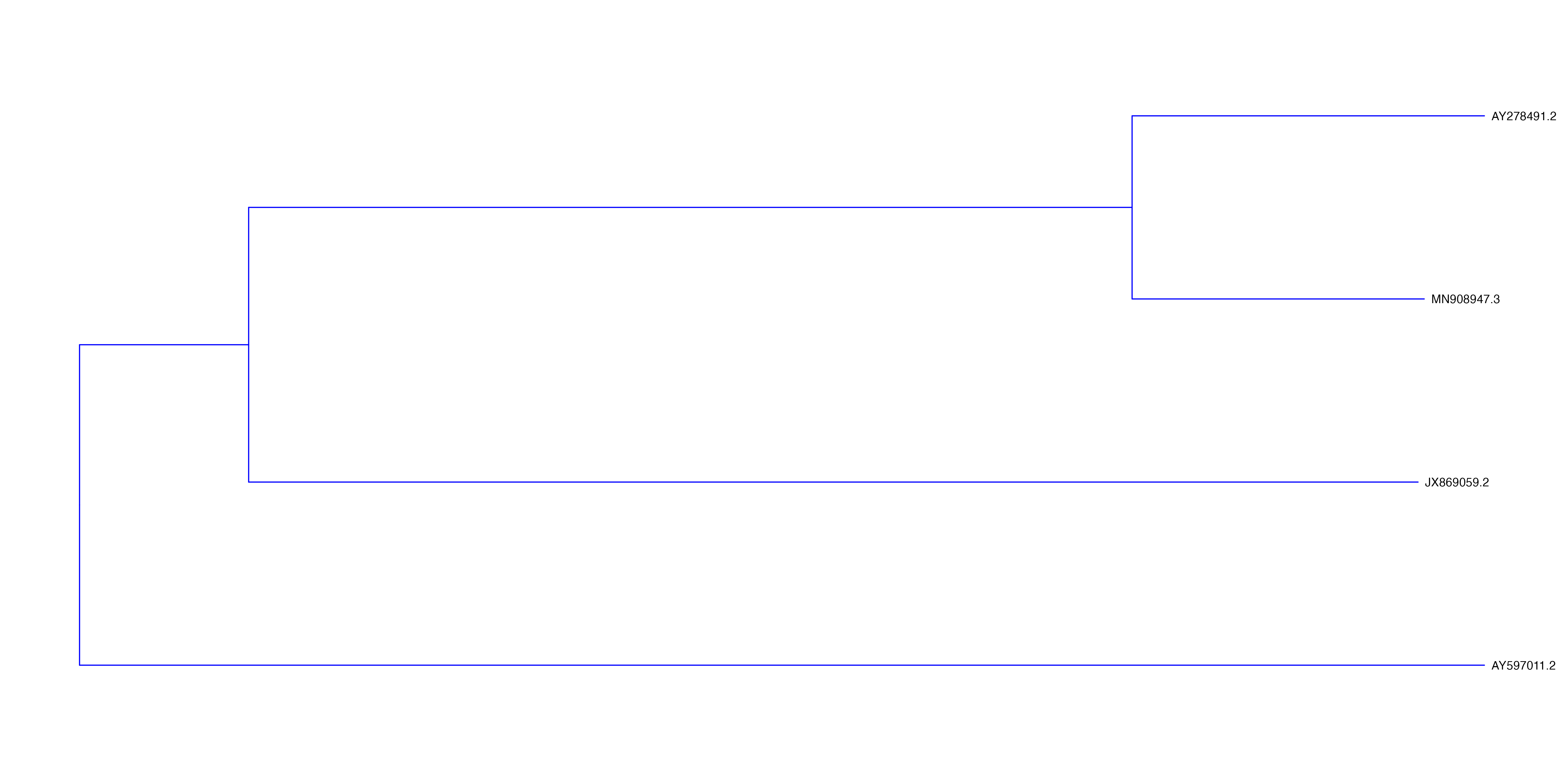
Looks quite good. But we can do better.
-
You can add more annotations to tree using metadata.
Prepare a metadata csv file like the following:
ID Name Genus Subgenus AY597011.2 HCoV-HKU1 Betacoronavirus Embecovirus JX869059.2 MERS-CoV Betacoronavirus Merbecovirus MN908947.3 SARS-CoV2 Betacoronavirus Sarbecovirus AY278491.2 SARS-CoV Betacoronavirus Sarbecovirus Note: Ensure one of the columns, in this case
IDmatches the names of the tree tipsImport this csv to R.
meta <- read.csv("./meta.csv") -
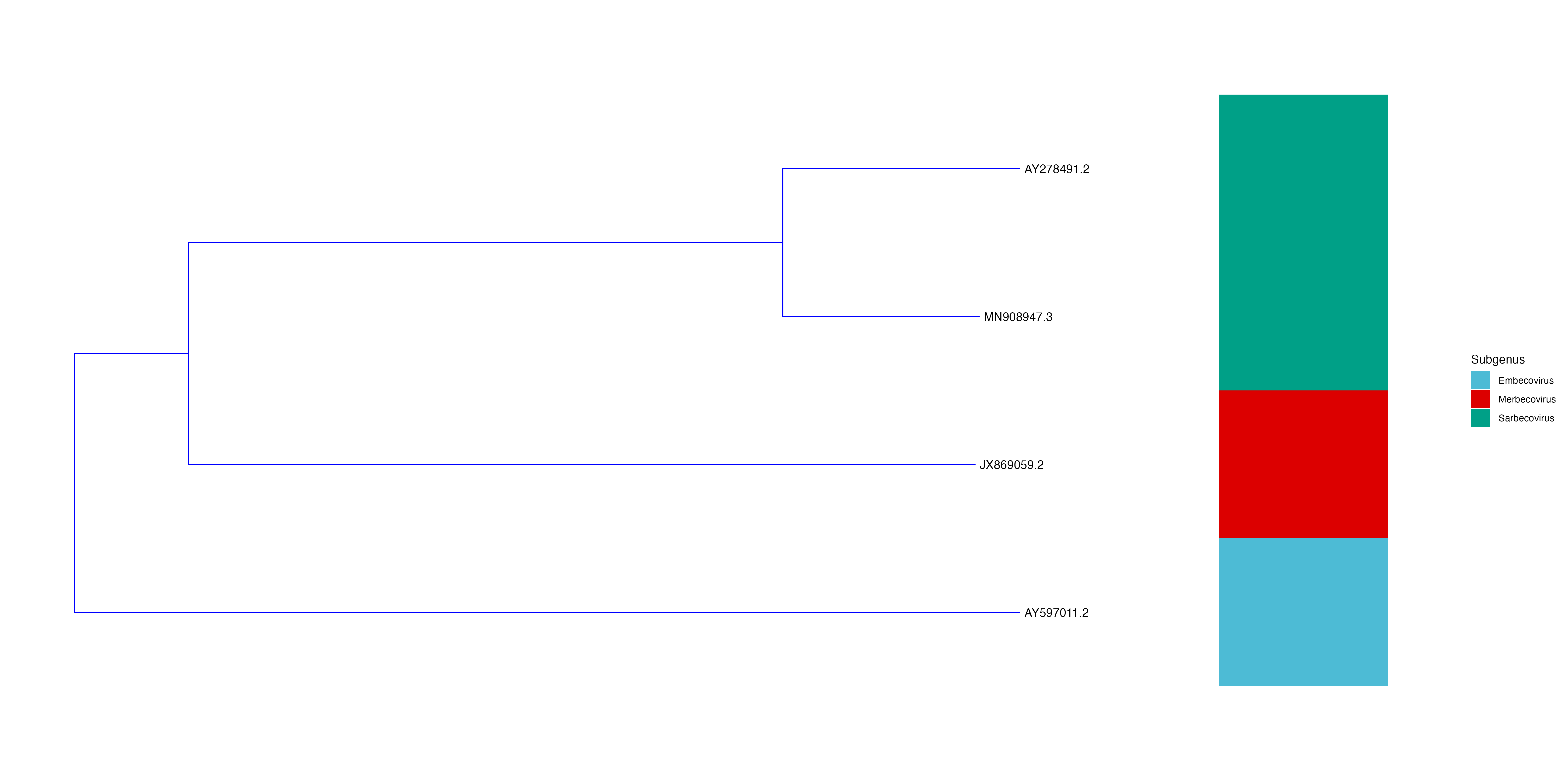
Now you can annotate the tree. In this example, the subgenus column is used.
p3 <- p2 + new_scale_fill() + geom_fruit(data = meta, geom = geom_tile, mapping = aes(y = ID, fill = Subgenus), offset = 0.3, pwidth = 0.1) + scale_fill_manual( values=c("#4DBBD5FF", "#DC0000FF", "#00A087FF"))Note: adjust the
offset,pwidthand colours as defined inscale_fill_manualaccording personal taste -
Plot and save
plot(p3) ggsave("./lineage.png", p3, width = 20, height = 10)