Marvelous Misadventures in Bioinformatics
A blog on some snippets of my work in bioinformatics. Hopefully you find something useful here and avoid stupid mistakes I made.
Compressed Sparse Row (CSR) Matrix
CSR is a very efficient way to store sparse matrices, such as one-hot vectors commonly found in machine learning tasks. This example compares the shiga toxin (Accession WP_095907599) one-hot encoded vector with a CSR matrix.
Prerequisite
- SciPy
- Numpy
- h5py
- Bio (optional)
Installation
pip install numpy
pip install scipy
pip install h5py
pip install biopython
Usage
-
Load libraries and dependencies
import sys import numpy as np from scipy.sparse import csr_matrix from Bio import SeqIO import h5py as h5Let’s also define some housekeeping variables
nucleotides = ['A', 'C', 'G', 'T'] -
Define a functions for one-hot encoding a sequence of nucleotides
def one_hot_naive(seq): #naive implementation arr = [] for s in seq: row = [0]*4 if s in nucleotides: row[nucleotides.index(s)] = 1 arr.append(row) return np.array(arr)The above is a naive implementation, but it’s not very pythonic nor efficient. Let’s use a better one.
def one_hot(seq): #more efficient implementation arr = np.zeros((len(seq), 4), dtype=int) for i, s in enumerate(seq): if s in nucleotides: arr[i, nucleotides.index(s)] = 1 return arr -
Load a nucleotide sequence
fastafile = "./stx1e_a.fna" #assuming you have a file called stx1e_a.fna in the same directory seq = [] with open(fastafile, "r") as readfile: for record in SeqIO.parse(readfile, "fasta"): seq.append(str(record.seq)) seq = seq[0] #we only have a single sequence, so this changes the list back to a string
OR
You could just hard code it into the script
#seq = 'ATGC...'The length of the sequence is 948 nucleotides.
-
One hot encode the sequence
one_hot_seq = one_hot(seq) one_hot_seq_naive = one_hot_naive(seq)We could check if both encoded sequences is the same
assert np.array_equal(one_hot_seq, one_hot_seq_naive)Nothing comes out when we run this, so both methods are one and the same.
-
CSR
To invoke csr compression, simply use the
csr_matrix()function. Note that we want to compress the one-hot vector.csr_one_hot_seq = csr_matrix(one_hot_seq)This will return a csr object, containing differnt attributes.
print(csr_one_hot_seq)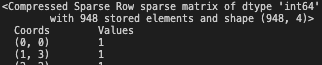
To access the data. We need to use
data,indices, andindptrattributes.print(f"Data = {csr_one_hot_seq.data}, dtype = {type(csr_one_hot_seq.data)}") print(f"Indices = {csr_one_hot_seq.indices}, dtype = {type(csr_one_hot_seq.indices)}") print(f"Indptr = {csr_one_hot_seq.indptr}, dtype = {type(csr_one_hot_seq.indptr)}")You will notice that all attributes are simply arrays.
The data attribute represents the actual “stuff” in the matrix, in this case, it’s all
1s because we are simply using one-hot encoding.
The indicies represent the position of the data in the matrix, in this case, it’s the index of the nucleotide in the sequence. Refer to the
nucleotideslist for the actual nucleotide.
The indptr attribute respresnts the mapping of data to the indicies. In this case, it’s a simple incremental array as each row only has 1 single non-zero value.
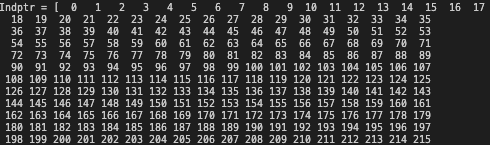
In cases where more than 1 column has non-zero values, the indptr may have duplicate indicies in the array.
-
Compare array sizes
We can compare the array byte size using
sys.getsizeof()print(sys.getsizeof(one_hot_seq_naive)) # 30464 bytes csr_size = (sys.getsizeof(csr_one_hot_seq.data) + sys.getsizeof(csr_one_hot_seq.indices) + sys.getsizeof(csr_one_hot_seq.indptr)) print(csr_size) # 4132 bytesWe can also save these arrays into files to compare actual disk usage.
with h5.File("csr_one_hot_seq.h5", "w") as f: f.create_dataset("data", data=csr_one_hot_seq.data) f.create_dataset("indices", data=csr_one_hot_seq.indices) f.create_dataset("indptr", data=csr_one_hot_seq.indptr) with h5.File("one_hot_seq.h5", "w") as f: f.create_dataset("one_hot", data=one_hot_seq)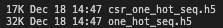
CSR compression saves about ~50% in size.
-
To regenerate the one-hot encoded sequence
To use the 3 arrays to regenerate the original one-hot encoded sequence, we can use the
csr_matrixfunction again.one_hot_seq_regen = csr_matrix((csr_one_hot_seq.data, csr_one_hot_seq.indices, csr_one_hot_seq.indptr)) one_hot_seq_regen = one_hot_seq_regen.toarray() #this is important to convert to a numpy array assert np.array_equal(one_hot_seq_regen, one_hot_seq)Nothing comes out so the one-hot vector has successfully been regenerated.
References
- https://docs.scipy.org/doc/scipy/reference/generated/scipy.sparse.csr_matrix.html