Marvelous Misadventures in Bioinformatics
A blog on some snippets of my work in bioinformatics. Hopefully you find something useful here and avoid stupid mistakes I made.
Multiple Sequence Alignment
Multiple sequence alignment (MSA) is a integral part of bioinformatics where two or more nucleotide or protein sequence can be aligned together. This MSA can be used to infer phylogenetic trees and the evolutionary relationships between sequences.
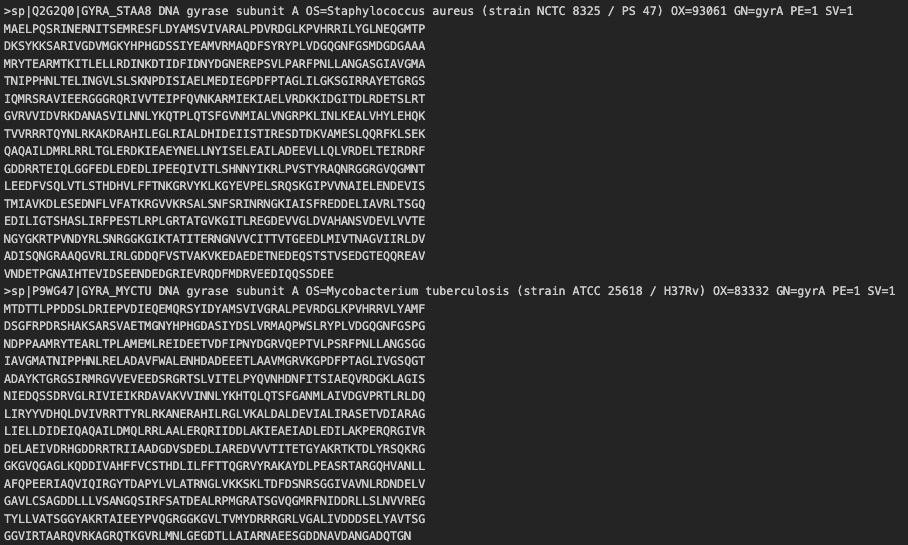
This small tutorial will use two bacterial gyrase subunit A (gyrA) protein sequences from M tuberculosis ATCC 25618 (Accession: P9WG47) and S. aureus NCTC 8325 (Accession: Q2G2Q0) by using the MAFFT aligner.
Prerequisite
- MAFFT
Installation
conda install -c bioconda mafft
Preprocessing
Ensure your sequences are in a multi-fasta format (i.e.)
>header
AAAAAAACCCCDDDD
>header
AAAACCCCDDDD
Usage
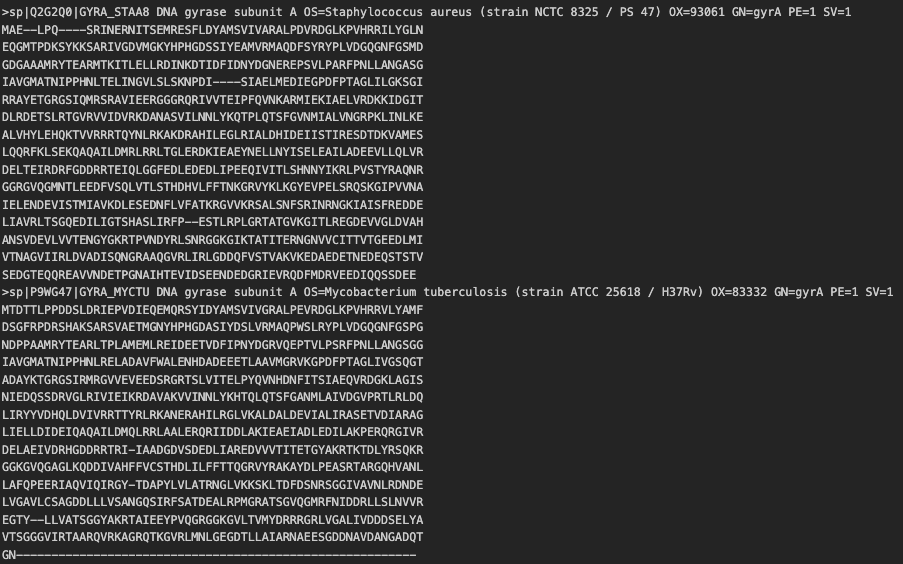
Assuming your multi-fasta file is called msa.fa, run the following command
mafft --auto --thread 1 msa.fa > msa.out.fa
where msa.out.fa is the output alignment file. The output also follows a multifasta format, but with misalignments between sequences marked with a -
Notes
--thread <int>controls how many threads to run mafft on, for larger sample sizes, enter a larger number.>redirects the fasta file into the file after the symbol.